Indice Anterior Siguiente
Revista de Ciencias Médicas La Habana 2014; 20(2)
PRESENTACIÓN DE CASO
Julio César Lima Regalado,I Jacinto Pedroso Caballero,II Dianelys Padín Martínez,III Anilexis Acosta BousoIV
IEspecialista de I grado en Ortopedia y Traumatología. Profesor Instructor. Máster en Urgencias Médicas. Hospital General Docente “Aleida Fernández Chardiet”. Güines, Mayabeque, Cuba. E-mail:dianelys.padin@infomed.sld.cu
IIEspecialista de I grado en Ortopedia y Traumatología. Profesor Instructor. Hospital General Docente “Aleida Fernández Chardiet”. Güines, Mayabeque, Cuba. E-mail:jacintop@infomed.sld.cu
IIIEspecialista de I grado en Cirugía General. Profesor Instructor. Máster en Urgencias Médicas. Hospital General Docente “Aleida Fernández Chardiet”. Güines, Mayabeque, Cuba. E-mail: dianelys.padin@infomed.sld.cu
IVMédico general. Hospital General Docente “Aleida Fernández Chardiet”. Güines, Mayabeque, Cuba. E-mail: anilexysab@infomed.sld.cu
RESUMEN
El Síndrome del hombre de piedra o miositis osificante progresiva es una enfermedad congénita del tejido conectivo con una frecuencia de 1 por cada 2 millones de nacidos vivos, es una enfermedad rara. Se reporta un paciente masculino, de 40 años, que acude a la consulta externa del Hospital General Docente “Aleida Fernández Chardiet”, del municipio Güines, provincia Mayabeque, por presentar dolor muscular, rigidez en los muslos, dificultad para deambular y mantener la posición de pie. Se decidió el ingreso en el servicio de ortopedia para realizar los estudios correspondientes. En los estudios complementarios se observa en las radiografías, osificación en los músculos glúteo mediano y vastos externos de ambas caderas sin antecedentes de trauma, y anemia en la hemoquimica. Se discuten hallazgos, se revisa literatura y se comparan las características de este paciente con las encontradas en otros casos, se llega al diagnóstico de fibrodisplasia osificante progresiva por mutación de novo, dado que ningún otro integrante de la familia ha sido afectado.
Palabras clave: osificación heterotópica, miositis osificante, fibrodisplasia osificante progresiva.
ABSTRACT
The stone man syndrome or myositis ossificans progressiva is a congenital connective tissue disease with a frequency of 1 in 2 million live births, it is a rare disease. It is reported a 40-year-old-male patient who attended the external consultation of “Aleida Fernández Chardiet” General Teaching Hospital of Güines municipality, Mayabeque province, for presenting with muscle pain, stiffness in the thighs, difficulty to wander and maintaining the standing position. It was decided the admittance into the service and the corresponding studies in relation to the patient. In complementary studies it was observed on radiographs, ossification in the gluteus medius and vastus externus muscles of both hips with no antecedents of trauma, and anemia in hemochemical. Findings are discussed, literature is reviewed and the characteristics of this patient with those found in other cases are compared, arriving at the diagnosis of fibrodysplasia ossificans progressiva due to a de novo mutation, since no other family member has been affected.
Key words: heterotopic ossification, myositis ossificans, fibrodysplasia ossificans progressiva.
INTRODUCCIÒN
La miositis o fibrodisplasia osificante progresiva (FOP) es una afectación rara. Se han descrito menos de 150 pacientes en EEUU, con una prevalencia de 1:2 millones de población y de carácter hereditaria1 se estima que afecta unas 2500 personas en todo el mundo.
Se debe a una mutación en el gen ACVR1 ubicado en el cromosoma 2, con un patrón de herencia autosómica dominante, aunque esporádicamente puede ocurrir por una mutación de novo, caracterizada por presentar al nacimiento: hallux valgus, microdactilia y monofalagnes en el I dedo de los pies, que se puede acompañar de microdactília en los pulgares, posteriormente osificación heterotópica, que restringe progresivamente el movimiento de las articulaciones, también puede aparecer de manera espontánea en cualquier momento de la vida, siendo más frecuente antes de los 10 años.1
Ocasionalmente se acompaña de calvicie, hipoacusia, osteocromatosis y retraso mental leve. Conocida también como miositis osificante progresiva que se cambió, en el año 1970, a fibrodisplasia, denominación que abarca la afección de más tejidos blandos además del músculo. Normalmente se presenta a edades tempranas, en niños, adolescentes o adultos jóvenes, siendo difícil establecer su diagnóstico en las primeras etapas de la enfermedad. La malformación del primer metatarsiano de los dedos de los pies es un signo inicial e importante.
Progresivamente se producen osificaciones heterotópica de los tendones, ligamentos, fascias y músculo estriado que afectan a estas estructuras en regiones próximas a columna vertebral, caderas y extremidades.Usualmente aparece afectación axial de progresión en sentido cráneo-caudal y proximal-distal.1,2
Presenta una evolución invariablemente progresiva, que confina a permanecer en una silla de ruedas a un alto porcentaje de pacientes sobre la 3ra década de vida y, a menudo, a padecer complicaciones pulmonares por insuficiencia respiratoria sobre la 5ta-6ta década.1-3
No existe un pilar fundamental en el manejo de esta enfermedad; ya que actualmente no hay un tratamiento médico eficaz. Por lo poco frecuente, se presenta un caso de paciente afectado de síndrome de hombre de piedra con osificaciones musculares progresivas y acompañado de invalides marcadas.
PRESENTACIÓN DEL CASO
Una vez obtenido el consentimiento informado, autorizado por la representante, se procedió al estudio de un paciente masculino de 40 años de edad que acudió al cuerpo de guardia del Hospital General Docente “Aleida Fernández Chardiet” del municipio de Güines, provincia Mayabeque, por presentar mialgia generalizada, a predominio de los miembros inferiores de moderada a intensa, de 15 años de evolución, de manera espontánea y sin causa aparente que lo desencadene. El nacimiento del paciente fue mediante parto eutócico, intrahospitalario, a las 40 semanas de gestación, con un peso al nacer de 4.0 kilogramos, tuvo excelente desarrollo psicomotor.
Los antecedentes maternos y paternos son nulos, sin consanguinidad. Presenta noveno grado de escolaridad, la que alcanzó luego de grandes esfuerzos, ya que presenta retraso mental. Al examen físico presenta fenotipo longilineo, conmucosas húmedas e hipocoloreadas, ruidos rítmicos de buen tono no soplo TA: 120/70 mmHg, pulso de 88 latidos por minutos, frecuencia respiratoria 18 respiraciones por minutos. Abdomen normal. Ojos, orejas, pupilas; conductos auditivos, fosas nasales, así como dentadura, sin alteraciones.
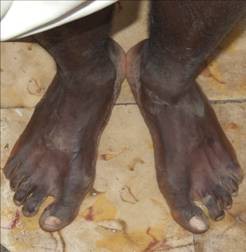
En el sistema osteomioarticular se observa Hallux Valgus interfalangico en el primer dedo de los pies (figura 1), pero sin microdactilea. Se palpa endurecimiento de ambas caras laterales de los muslos hasta las rodillas, dificultad para la marcha por dificultad al extender ambas rodillas y la fuerza muscular es 3 al extender las rodillas.
Fig. 1. Hallux Valgus interfalángico de ambos gruesos artejos
Además presenta maniobras positivas de cadera izquierda dado por Patrick positiva, así como Thomas. En los estudios que se le realizaron al paciente se encontró que solo en la hemoquímica se encontraba anemia de 90 gramos por litros, y la elevación de las cifras de la fosfatasa alcalina 550 unidades por litro, indicativa de actividad de la enfermedad y consecuencia de la existencia de un aumento del recambio óseo.
En los estudios radiográficos la osificación de ambos vastos externos (figura 2), así como los músculos glúteos mediano y mínimo izquierdos (figura 3) son los hallazgos encontrados. No presenta osificaciones de otros grupos musculares del cuello ni regiones de la columna vertebral.
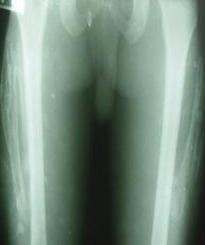
Fig 2. Osificación de ambos vastos externos
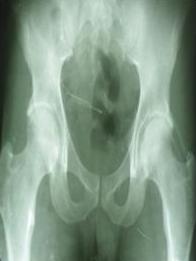
Fig. 3. Osificación de músculos glúteo mediano y mínimo izquierdos
Ingresado en el servicio de ortopedia se le instauró después de los complementarios en la hemoquímica, ibuprofeno 400 miligramos cada 8 horas, así como dipirona 600 miligramos cada 6 horas si dolor. No se le realizó estudios de biopsia.
DISCUSIÓN
La FOP es causada por una mutación del gen ACVR1 que codifica para un receptor denominado ACVR1 en la vía de señalización de la proteína morfogénica ósea (BMP), que se expresa en diversos tejidos incluyendo el músculo esquelético y condrocitos.1,3,4 En la fibrodisplasia osificante progresiva una línea linfoblastoide de leucocitos sobreexpresan una proteína específica productora de hueso (proteína morfogenética 4 o BMP4) y subexpresan proteínas supresoras de la BMP4 (proteínas noggin y gremlin).
La BMP4 atrae células mononucleares que comienzan el proceso de angiogénesis, estimulan la formación de tejido fibroso de células madre mesenquimales y la apoptosis, provocando un proceso que resulta en la formación de trozos de hueso heterotópico maduro que reemplaza el tejido músculo-esquelético y demás tejido conectivo. El hueso heterotópico maduro en estos pacientes es bioquímica e histológicamente indistinguible del hueso esquelético normal.1-4
Los niños con FOP parecen normales al nacer, excepto por la malformación del I dedo de los pies, que en el caso estudiado presentaba hallux valgus interfalangico. Durante la primera o segunda década de vida, a los niños se les forman dolorosos nódulos fibrosos en el cuello, espalda y hombros, los cuales son precedidos de edema duro con signos de flogosis, dichos nódulos crecen como hueso, en un proceso conocido como osificación heterotópica.
Luego progresa a lo largo del tronco y las extremidades. Este fenómeno se puede agravar y progresar con extensión de la osificación a cadera y extremidades, siendo el paciente presentado algo atípico porque comenzó por estas últimas regiones, denominándose los casos descritos en la literatura como “hombre de piedra”.1,5
Estas lesiones lentamente reemplazan los músculos del cuerpo con hueso aparentemente normal, como característica los músculos del diafragma, lengua, ojos, cara y corazón no son afectados.2,4,5 Cualquier intento de remover los huesos adicionales da como resultado una formación ósea, aún más robusta.
Entre los diagnósticos diferenciales para la FOP se tiene el cáncer, las lesiones FOP pueden aparecer repentinamente y producen inflamaciones severas en pocas horas. Esto desconcierta a los oncólogos por la expansión y crecimiento repentino de un tumor, sin saber su origen. Para averiguarlo se sugiere una biopsia de diagnóstico. Una lesión FOP temprana se puede confundir con diferentes tipos de cáncer dependiendo del estado de maduración de la lesión así como con lesiones traumáticas.5,6
La razón para esto es que cuando se forma un nuevo hueso a través de un proceso endocondral (como lo hace el hueso FOP), pasa por varias etapas de proliferación celular, incluyendo células que están involucradas en el tejido conectivo, cartílago y hueso.
Sería más fácil realizar un diagnóstico de FOP, si al tener sospecha de ésta se correlaciona con las características anatómicas de los dedos del paciente; así como el crecimiento más rápido de las lesiones que cualquier tipo de cáncer.
En segundo lugar la fibromatosis infantil agresiva (FIA) donde los fibroblastos proliferan en varios tejidos, incluyendo músculo, tendón, ligamento. Esta podría confundirse si se realiza una biopsia de una lesión de FOP, debido a que en fases tempranas ambas lesiones lucen idénticas, sin embargo, las lesiones de FIA no progresan más allá de la fase del crecimiento del tejido conectivo, mientras que en FOP maduran para formar cartílago y hueso.
Una vez que se observan los cartílagos o células óseas en una lesión, ya no se deberían confundir, además, los pacientes con esta enfermedad tienen sus dedos deformados, lo cual serviría para descartar dicho diagnóstico.5-7
Debido a la naturaleza genética de la FOP existe un 50 % de probabilidad que los hijos de un afectado herede la mutación. Aunque es posible para las mujeres con FOP concebir y tener hijos, es peligroso y atenta contra su vida.
La FOP no mejora con el tiempo, sino que empeora mientras envejezca el paciente. La mayoría de los pacientes terminan postrados en silla de ruedas a los 30 años aproximadamente y la muerte llega por la restricción del tórax, que causaría insuficiencia respiratoria.8
Aunque se trata de una combinación única de anomalías esqueléticas y osificaciones ectópicas, el diagnóstico inicial es a menudo erróneo hasta en un 90 %. Las imágenes radiográficas suelen no mostrar alteraciones hasta después de 6 a 12 meses que aparece el edema, momento en que se observan depósitos de calcio y comienza la afectación, generalmente por el omóplato y columna cervical.
La biopsia no es necesaria para su diagnóstico y debe ser evitada para no desencadenar nuevos focos de osificación, parámetro que se tuvo en consideración. Siendo de esta forma el diagnóstico eminentemente clínico e imagenológico.1,6,9
No existe tratamiento efectivo para la FOP. El etidronato disminuye la osificación de nuevos nódulos, pero no influye sobre las lesiones antiguas osificadas. También se ha usado berilio, vitamina B, vitamina E, corticosteroides, antiinflamatorios y penicilamina. Ninguno de estos medicamentos fue efectivo.
El manejo de pacientes con FOP, debe ser dirigido fundamentalmente a la protección del niño, evitando traumatismos, cirugías, biopsias e inyecciones intramusculares. En lo que se refiere al tratamiento, al tratarse de una patología de base genética, no existe tratamiento etiológico ni se ha encontrado ningún tratamiento realmente efectivo.
Solo es posible recomendar medidas encaminadas a minimizar los riesgos de lesión como la limitación de actividades que puedan activar un nuevo brote. La afección evoluciona comúnmente por brotes, alternando con épocas quiescentes que alteran la autonomía del paciente, hasta que el desarrollo de una incapacidad grave o una insuficiencia respiratoria secundaria a una restricción del tórax le ocasionan la muerte.2,5,8,10
REFERENCIAS BIBLIOGRÁFICAS
Recibido: 11 de diciembre de 2013.
Aprobado: 3 de febrero de 2014.
Dr. Julio César Lima Regalado.Especialista de I grado en Ortopedia y Traumatología. Profesor Instructor. Máster en Urgencias Médicas. Hospital General Docente “Aleida Fernández Chardiet”. Güines, Mayabeque, Cuba. E-mail: dianelys.padin@infomed.sld.cu