Dra. Lourdes M. Moreno Pérez1, Dr, Felipe Herrera Ramos2, Téc. Ediel Peraza Martínez3.
Colaboradora: Romina Herrera Moreno
Estudiante de 6to año de Medicina.
Se reporta la presencia de una familia portadora del síndrome de Klein-Waardemburg en el municipio de Artemisa existiendo varios miembros afectados con diversos grados de expresividad en las tres últimas generaciones de esta enfermedad infrecuente, autonómica dominante que cursa con cierto grado de discapacidad cuando aparece la hipoacusianeurosensorial. No se pudo clasificar a los enfermos según las variantes del Síndrome, pues los mismos carecían de estudios genéticos. Se expone además una revisión bibliográfica de los aspectos genéticos que dan lugar a la aparición del síndrome.
Descriptores DeCS: SINDROME DE WAARDENBURG/genética; SINDROME DE WAARDENBURG/fisiopatología; IRIS; SORDERA
El síndrome de Klein-Waardemburg o síndrome de Fijación Embrionaria fue descubiertó en 1951 y se define como una rara enfermedad autosómica dominante con penetrancia variable y varios tipos clínicos conocidos (WSI, WSII, WSIII y WSIV) teniendo una incidencia de 1 en 40000 habitantes y un 50% de riesgos de heredar la enfermedad los hijos de progenitores enfermos; no así los miembros sanos los cuales no transmiten la enfermedad a sus descendientes.
Ambos sexos tienen la misma probabilidad de transmitirlos1,2. Este síndrome se caracteriza por presentar signos oculares y generales. Dentro de los primeros se encuentran la hiperplasia de la porción interna de las cejas, el aumento de la distancia interpupilar, hipodesarrollo de las órbitas, desplazamiento lateral de los ángulos internos y puntos lagrimales, blefarofímosis, hipoplasia de la carúncula, engrosamiento del tarso, epicanto, y heterocromía del iris.
Dentro de los signos generales, se puede encontrar: hipoacusianeurosensorial, Raíz nasal ancha y alta, Braquicefalia, Cabello albinótico, Albinismo parcial, y Anomalía en las extremidades superiores 3,4. Se destacan en negritas los rasgos más característicos.
Características genéticas: La enfermedad es causada por un desarrollo defectuoso de la Cresta Neural (Neorocrestopatía), lo que conlleva a una alteración en la función de la misma. Este Síndrome presenta cuatro variantes en dependencia de la alteración genética y de la presencia o ausencia de síntomas adicionales1,5.
El Síndrome de Klein-Waardemburg tipo I y tipo III son debidos a mutaciones en el gen PAX3 (2q,35)5-8. Lo característico del tipo I es la DistopiaCanthorum9. Lo del tipo III es una Deleción Heterocigótica en el gen PAX3 caracterizado por grandes anomalías músculo-esqueléticas en asociación con rasgos faciales comunes con el tipo I.
En un estudio realizado en Bélgica se reporta una familia con lesión Heterocigótica 13-bp en el par dominante del gen PAX310.
El Síndrome de Klein-Waardemburg tipo II es heterogéneo estando causado por mutación en el factor de transcripciónmicroftalmía asociada (MITF), esencial para la diferenciación melanocitica. La alteración específicamente está en la región 8p23 (Gen OC2) lo que se demuestra que el OC2 estimula la expresión del MITF siendo este un gen candidato y no causa común del síndrome. Este tipo presente alteraciones sensoriales del oído por la ausencia de melanocitos6,11. También se han detectado translocaciones en el cromosoma (4p..8p)12 y de lesiones homocigóticas en SLUG responsables de los síndromes pigmentarios6,13.
El tipo III, llamado también SHAH- Waardemburg Síndrome, resulta de mutación en el gen endotelial beta receptor (EDNRB), en el gen EDN3 o en el gen SOX10. Este último, en sinergismo con el PAX3 activan fuertemente al MITF 5,14,15,16. Mutaciones en SOX10 (Q250X) produce un fenotipo de WaardemburgHirschsprung en el cual se acompaña de neuropatía y desmielinización central por anomalía en las células de SCHWAMN17.
Otro estudio realizado en Turquía a un niño de dos meses de edad se le detectaron dos translocaciones separadas; una entre el cromosoma 1 y 8 y otra más compleja entre 4 y 718. Se ha reportado también mutación en el cromosoma 2q como causa del síndrome en estudio19. Podemos decir que en este síndrome existe una cascada de genes que pueden ser los responsables de la enfermedad (PAX3, MITF, HUMAN MYOD, MYF5, C-MET, C-KIT, TYROSINASE, TRP-1, HUMAN QNR-71, SOX10, EDNRB y EDN3) por lo que mutaciones de diferentes genes pueden expresar la enfermedad 20.
En este trabajo se presenta una familia portadora de este síndrome infrecuente en nuestro medio, con el objetivo de constatar las implicaciones médico sociales y el grado de discapacidad asociado a estos enfermos, y su enfrentamiento eficaz por el nivel de atención primaria de salud en nuestro municipio.
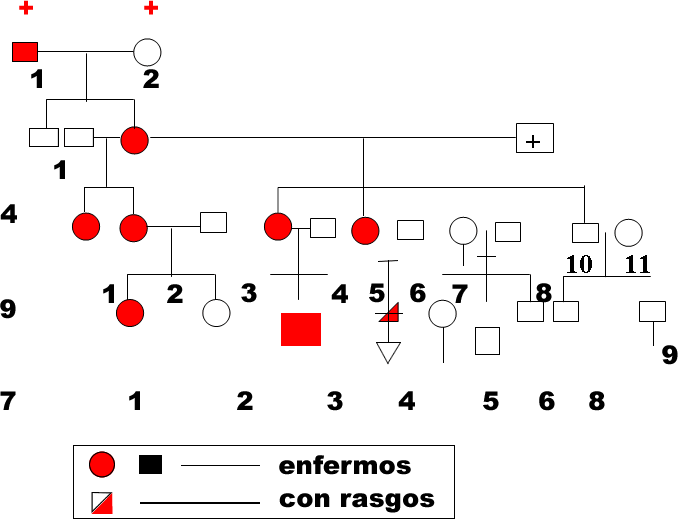
Se trata a una familia residente en el poblado de Neptuno, municipio de Artemisa. Esta familia cuenta de varios miembros afectados de la enfermedad, la cual se ha presentado en varias generaciones. Existe una limitante en este caso y es que la anciana madre de los enfermos no recuerda las características de su ascendencia. Para ejemplificar el predominio dominante de la enfermedad, observe en el árbol genealógico de esta familia (gráfico 1) la expresión de los rasgos de la enfermedad, destacándose las diferentes formas de expresividad en cada uno de los miembros de esa familia y en sus distintas generaciones.
Gráfico 1. Árbol genealógico
El Síndrome de Waardemburg es una enfermedad infrecuente que provoca alto grado de discapacidad cuando se acompaña de hipoacusia. Para su mejor estudio se divide la sintomatología en cuatro grupos:
Dentro de los rasgos dismórficos los más frecuentes son: Hiperplasia de la porción interna de las cejas, desplazamiento lateral de los ángulos internos y puntos lagrimales, raíz nasal ancha y alta, braquicefalia, y aumento de la distancia interpupilar. Con relación al mechón blanco es un signo de albinismo parcial localizándose la hipopigmentación en áreas pequeñas21.
La Heterocromía del iris puede ser parcial o total o puede no aparecer en dependencia de la expresividad del gen. La sordera es una hipoacusia profunda neurosensorial, incluida dentro de la sordera genética sindrómica, representando el 40% de las mismas23. Un estudio realizado en Oregon Estados Unidos demostró que esta enfermedad cursa con alteraciones de la función vestibular con más incidencia que con sordera demostrada por alteraciones electrococleográficas24.
En la tabla 1 se muestran las diferentes formas de expresividad del síndrome en los enfermos. Como se observa, esta afección cursa con diversidad de expresión existiendo personas oyentes y personas sordas, las cuales necesitan de una educación individualizada debido a los graves problemas que genera en el entorno la falta de comunicación y necesidades de aprendizaje, generando una limitación en su interacción emocional, intelectual y física25.
Tabla 1- Formas de presentación de los diferentes rasgos en los pacientes.
PACIENTE |
RASGOS DISMÓRFICOS |
MECHÓN BLANCO DEL CABELLO |
HETEROCROMIA DEL IRIS |
SORDERA NEUROSENSORIAL |
MADRE |
X |
X |
X -TOTAL |
|
HIJA 1 |
X |
X |
X -TOTAL |
X |
HIJA 2 |
X |
X |
X -TOTAL |
|
HIJA 3 |
X |
|
X -TOTAL |
|
HIJA 4 |
X |
|
X -PARCIAL |
|
NIETO 1 |
X |
|
|
|
NIETO 2 |
X |
X |
|
|
| NIETO 3 | X |
X |
X |
|
Fuente: Árbol genealógico.
Es importante destacar, después de haber expuesto todas las variantes genéticas que pueden dar lugar a la aparición del síndrome, que se debe, ante estos casos, realizar un asesoramiento genético con vistas a erradicar una causa de discapacidad en el ser humano. En este sentido, se reportan experiencias de consejo genético en México26 y en Ámsterdam27 con mujeres embarazadas.
En este estudio no fue posible clasificar las variantes del síndrome en las personas afectadas pues carecían de estudio genético (cariotipo) en el momento de realizar la investigación de discapacitados en el municipio de Artemisa, por lo cual no se pudo profundizar en los aspectos genéticos de la familia.
Dada la repercusión social de este síndrome en la familia estudiada, es recomendable crear comisiones de trabajo con médicos, enfermeras, especialistas de ORL y Oftalmología con vistas a prevenir y detectar anomalías congénitas causantes de sordoceguera.
The presence of a carrier family of Klein-Waardenberg Syndrome in Artemisa municipality existing some members affected with diverse grades of expresivity in the last three generations of this infrequent disease,dominant autonomic which brings certain grade of incapacity where appears the neurosensorial hypoacusy was reported.We couldn`t classify our sicks according to the syndrome variants because they hadn`t any genetic study. We also point out a bibliographic revision of the genetic aspects which bring the syndrome starting.
Subject headings:WAARDENBURG´S SYNDROME /genetics; WAARDENBURG´S SYNDROME /physiopathology; IRIS; DEAFNESS