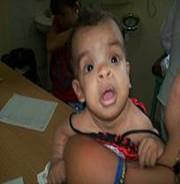
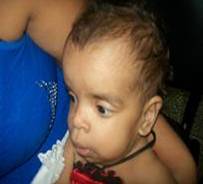
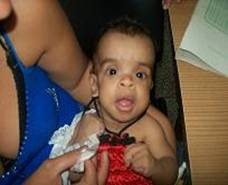
Figura 1. Características faciales de la afectada.
Medimay 2017; 24(1)
PRESENTACIÓN DE CASO
Elayne E. Santana Hernández,I Víctor Jesús Tamayo Chang,II Odette Wagner Vigo,III Onelis Pupo Zalazar.IV
IEspecialista de I y II grado en Medicina General Integral y Genética Clínica. Máster en Atención Integral al Niño. Asistente. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Holguín, Cuba. Correo electrónico: elsantana@infomed.sld.cu
IIEspecialista de I y II grado en Genética Clínica. Máster en Atención Integral al Niño. Profesor Auxiliar. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Holguín, Cuba. Correo electrónico vtamayo@hpuh.hlg.sld.cu
IIIEspecialista de I y II grado en Medicina General Integral y Genética Clínica. Instructor. Centro Provincial de Genética Médica de Holguín. Holguín, Cuba. Correo electrónico: owarner@hpuh.hlg.sld.cu
IVLicenciada. en Enfermería. Máster en Asesoramiento Genético. Instructor. Centro Municipal de Genética de Moa. Holguín, Cuba. Correo electrónico: onelisp@moa.hlg.sld.cu
RESUMEN
El síndrome de Costello es una enfermedad poco frecuente, se caracteriza por talla baja, retraso en el desarrollo y facies característica. Se hereda con patrón autosómico dominante, aunque se han descrito muchos casos esporádicos, donde la mutación se produjo en células germinales, apareciendo de novo, como mutación nueva. Se presenta un caso con retardo en el desarrollo pondoestatural y psicomotor, asociado a facie tosca. Se considera importante el método clínico para realizar diagnóstico de estas enfermedades raras, donde es necesario la estimulación temprana y el asesoramiento genético a los familiares
Palabras clave: síndrome de Costello; anomalías craneofaciales; insuficiencia del crecimiento; enfermedades raras.
ABSTRAC
Costello Syndrome is not a very frequent disease, it is characterized by low size, delaying in the development and characterized facies. It is inherited with dominant autosomic pattern, although many sporadic cases have been described, where the mutation was produced in germinal cells appearing again, like a new mutation. A case with delaying in the size and psychomotor development associated to facie tosca was presented. The clinical method was considered important for diagnosing these rare diseases, where the early stimulation and the genetic councelling to relatives was necessary.
Keywords: Costello syndrome; craniofacial abnormalities; failure to thrive; rare diseases.
INTRODUCCIÓN
El síndrome de Costello es descubierto por el Dr. Jack Costello, un pediatra de Nueva Zelanda en 1977, en dos sujetos con buen peso al nacer y dificultades en la alimentación en la vida postnatal con retraso del crecimiento, facies tosca y anomalías cardiacas. Se desconoce la prevalencia mundial ni por regiones por la poca documentación en la literatura.1,2
Los signos clínicos como los dermatológicos incluyen piel redundante en el cuello, palmas, dedos y en las plantas del pie (con hiperqueratosis de las palmas y de las plantas del pie, y engrosamiento de una piel fláccida en brazos y piernas), acantosis nigricans y papilomas.3
El déficit de crecimiento durante los primeros meses de vida conlleva a una estatura baja a pesar del aumento normal de peso en etapas posteriores. Es frecuente que se acompañen de cardiopatía congénitas.4 Presentan déficit intelectual grado variable, aunque suelen por lo general ser sociable y amigables.3 La hiperextensibilidad de los dedos es frecuente y en ocasiones presenta rabdomiosarcomas.5
La mayoría de los casos están causados por mutaciones de novo de la familia ras de oncogenes, HRAS.4 El diagnóstico depende principalmente del cuadro clínico: los papilomas representan la manifestación más característica, pero puede presentarse en etapas posteriores de la vida. El peculiar curso de la enfermedad, facie típica, y la afectación ectodérmica con piel fláccida e hiperpigmentada son características suficientes para hacer el diagnóstico.6-8 El diagnóstico diferencial debe incluir el síndrome de Noonan y el síndrome cardio-facio-cutáneo. No hay un tratamiento específico para el síndrome de Costello, sin embargo el diagnóstico precoz permite implementar medidas encaminada a la estimulación temprana mejorando el desarrollo psicomotor así como una adecuada nutrición.
Tambien se deben realizar exploraciones cardiacas para identificar defectos del corazón, y en correspondencia a con esto indicar terapia física a través de la terapeuta y rehabilitadores. El pronóstico depende de la gravedad de la cardiomiopatía y de la presentación de tumores malignos.
Se presenta este caso poco frecuente, por la importancia de realizar un diagnóstico oportuno para la estimulación temprana, encaminada a mejorar la calidad de vida de estos enfermos y su adecuada incorporación social, así como brindar apropiado asesoramiento genético a la familia.
PRESENTACIÓN DE CASO
Paciente femenina YSM de 14 meses de edad, que asiste a consulta de Genética Clínica remitida de neurodesarrollo al Centro Provincial de Genética de Holguín, porque a partir de los 4 meses de edad mantuvo peso estacionario, no rotaba sobre el abdomen y en su área el pediatra diagnosticó, un ligero retardo en el desarrollo psicomotor y pondoestatural, además de facie tosca y la envía a consulta neurodesarrollo.
En el interrogatorio no se refieren antecedentes patológicos de interés, no consanguinidad. Historia obstétrica: embarazo 1, parto 1, aborto 0. Tampoco se recogen antecedentes prenatales. Los antecedentes perinatales: parto eutócico a las 39,3 semanas, peso: 2280 gramos, talla: 47 cm, circunferencia cefálica (CC): 34 cm, apgar 8-9. Diagnosticándose al nacimiento un crecimiento intrauterino retardado.
En consulta de neurodesarrollo a los 4 meses, fue valorada por un equipo multidisciplinario formado por especialista de Neurología, Pediatría, Fisiatría, Nutrición, donde se efectúa valoración ponderal, se encontró la relación peso/edad: 4250 gramos por debajo del tercer percentil y talla/edad: 57cm por debajo del tercer percentil, peso y talla por debajo del tercer percentil, no sostenía la cabeza, ni tampoco rolaba sobre el abdomen. Se establece una dieta para mejorar su estado nutricional y se solicita valoración por cardiología que después de realizarle ecocardiograma se confirma que no tiene ninguna alteración cardiovascular. Se recomienda comenzar la estimulación temprana y rehabilitación muy favorable en estos casos para alcanzar un desarrollo psicomotor acorde a su edad.
En consulta de Genética Clínica, se definen las características siguientes: facie tosca, cráneo con tendencia a la braquicefalia, escaso cabello, hendiduras palpebrales amplias, puente nasal plano ancho con nares antevertidas, filtrum largo, lengua que la mantiene por fuera de los labios gruesos y evertidos. Se decide esperar el actuar de las recomendaciones anteriores y se cita a consulta en 3 meses.
En consulta a los 7 meses, se realiza valoración ponderal, se comprueba que se mantienen por debajo del tercer percentil los parámetros peso/edad: 5990 gramos, talla/edad: 62 cm, y CC: 44 cm en el 75 percentil, aún no rola sobre el abdomen, todavía sin control cefálico, no se sostiene sentada con apoyo. Mantiene facie tosca, con cráneo braquiocefálico, frente amplia abombada con implantación alta del cabello, región superciliar prominente y hendiduras palpebrales horizontalizadas, puente nasal plano ancho con nares antevertidas, hipertelorismo, orejas grandes en rotación posterior, filtrum largo con labios gruesos (figura 1), asociado a fallo de medro y retardo motor, se discute en colectivo lográndose delinear bien el fenotipo, se llega al diagnóstico clínico del Síndrome Costello.
Figura 1. Características faciales de la afectada.
Se le solicita a los padres consentimiento informado para tomarle fotos para comparar con otros casos similares y publicar en revistas científicas, respetando los principios éticos para investigaciones médicas.
Se brinda asesoramiento genético a los familiares entregando hoja informativa donde se explica todo lo referente a esta enfermedad, haciendo referencia el carácter hereditario de esta afección donde con alta frecuencia aparecen casos nuevos de forma esporádica como lo ocurrido en esta familia sin antecedentes de ningún otro afectado, pero por lo general, presentan un patrón de herencia autosómico dominante y el riesgo es alto de que lo transmita a su descendencia.
Continua en consulta multidisciplinaria de neurodesarrollo y de genética clínica con vigilancia nutricional y ponderal trimestral, ultrasonido abdominal, ultrasonido del sistema nervioso central mantiene fontanela anterior abierta, continua con la estimulación temprana y la rehabilitación para mejorar la hipotonía de tronco.
En consulta a los 9 meses se aprecia que la lactante mantiene iguales características faciales, pero sostiene la cabeza, gira sobre el abdomen se mantiene sentada con apoyo, refiere la madre que sostiene objetos con ambas manos y pronuncia monosílabas. Valoración ponderal peso/edad 6700 gramos, se encuentra en el tercer percentil, talla/edad 66,5 cm entre el tercero y décimo percentil, circunferencia cefálica CC: 46,5 entre el 90 y 97 percentil, se valoran los complementarios indicados donde ambos ultrasonidos son normales.
Se aprecia que después que mejoro nutricionalmente, avanzo en el desarrollo psicomotor, mantiene curva de crecimiento lento como lo esperado en estos casos, sin embargo es importante destacar la mejoría que alcanza con la estimulación temprana y la rehabilitación, la cual se mantiene para alcanzar mayor tono muscular que le permita la bipedestación antes de los 18 meses, lo que aún no ha logrado.
En consultas de asesoramiento genético se explica a los familiares, la importancia de no exponerla a radiaciones ni otro mutágenos que pueda causar mutaciones que produzcan la pérdida de heterocigocidad y para esto mantenemos vigilancia anual con ultrasonidos abdominales y otros estudios si fueran necesarios.
DISCUSIÓN
Se plantea que el Síndrome Costello (SC) es un raro y complejo desorden del desarrollo. Se caracteriza por la presencia de retraso del crecimiento postnatal, discapacidad intelectual de grado variable, facie típica: macrocefalia, implantación baja de las orejas, con lóbulos grandes y gruesos, labios gruesos; ventanas nasales antevertidas. Se describen estas características faciales asociadas a alteraciones dentarias1 que por la edad que tiene la afectada aún no se puede evaluar este aspecto.
Pueden presentar además pliegue nucal redundante, piel laxa con pliegues profundos en palmas, plantas y dedos, acantosis nigricans; hiperqueratosis palmoplantar, hiperlaxitud articular de los dedos, papilomas periorales y perinasales, asociado a enfermedades cardiovasculares y pulmonares que ensombrecen la esperanza de vida en estos casos.2 En la paciente se observa solamente acantosis nigrican en la nuca, sin poder evaluar la hiperlaxitud por la edad.
Típicamente, los síntomas faciales son groseros con frente ancha, epicantos, labios y lóbulos de las orejas gruesos. Aproximadamente el 63% de los pacientes tienen malformaciones cardiovasculares,1,2 lo que es diferente en esta paciente.
Por lo general los enfermos exhiben retardo del lenguaje, la memoria y trastornos cognitivos,3 que en este caso aun no son evaluables por la edad que tiene la afectada.
El 82 %-92 % de los pacientes con SC tienen una mutación germinal en el gen HRAS en el cromosoma 11p13.3, que codifica para un miembro de la superfamilia de las pequeñas proteínas de unión GTP. Las mutaciones resultan en un aumento de la función, es decir una ganancia de función, mediante la activación constitutiva de las señales en los mecanismos de control y diferenciación celular. El SC es causado por cualquiera de al menos cinco mutaciones diferentes en el gen HRAS en el cromosoma 11. Este gen proporciona instrucciones para hacer una proteína, H-Ras, que ayuda a controlar el crecimiento y la división celular.4
En lugar de desencadenar el crecimiento celular en respuesta a las señales particulares de fuera de la célula, la proteína hiperactiva dirige a las células para crecer y dividirse constantemente. Esta división celular sin control puede predisponer a los pacientes para el desarrollo de tumores benignos y malignos.5 Por lo que es muy beneficiosos mantenerse vigilante ante cualquier síntoma nuevo, evaluando los estudios ultrasonograficos y recomendando a la familia no exponer innecesariamente al afectado a radiaciones.7-9
El HRAS es un proto-oncogen en la que las mutaciones somáticas en las personas sanas pueden contribuir al cáncer. Considerando que los niños con SC típicamente tienen una mutación en HRAS en todas las células de sus cuerpos. Es primordial tener presente que si ocurre una segunda mutación con perdida de heterocigocidad, este proto-oncogen se encontraría en doble dosis es decir de forma homocigótica, y entraría en división celular de forma incontrolada desarrollándose el tumor maligno, lo que logra explicar por la teoría de Nudson. La prueba para la mutación en los tumores de cáncer también se puede usar para examinar a los niños para el SC,6 que en nuestro medio se realiza a través de estudios de alfa feto proteína en sangre periférica. En el caso que se presenta no se ha podido demostrar la mutación por estudios moleculares del gen HRAS, sin embargo otros autores han descrito casos con mutaciones comprobadas en este gen.10-12
Es importante realizar un diagnóstico lo más precoz posible para poder brindar adecuado asesoramiento genético a estas familias, realizar prevención primaria para disminuir la probabilidad de que se desarrollen tumores y poder mantener vigilancia sobre las posibles complicaciones que se pueden presentar en el curso de la enfermedad. Cuando se orienta adecuada a la familia se mejora el estado nutricional y se comienza tempranamente la rehabilitación, mejoran el desarrollo psicomotor y se propicia mejorar la calidad de vida de estos enfermos.
REFERENCIAS BIBLIOGRÁFICAS
Recibido: 30 de enero del 2016
Aprobado: 26 de noviembre del 2016
Elayne E. Santana Hernández. Especialista de I y II grado en Medicina General Integral y Genética Clínica. Máster en Atención Integral al Niño. Asistente. Investigador Agregado. Centro Provincial de Genética Médica de Holguín. Holguín, Cuba. Correo electrónico: elsantana@infomed.sld.cu